![]()
HIE-ISOLDE: a unique window into the nucleus
With the recent completion of the second phase of HIE-ISOLDE, CERN’s ISOLDE facility has a machine capable of answering long-unanswered questions about the nature of the nucleus. The installation of 4 cryomodules in 2018 allowed for the acceleration of radioactive isotopes up to 7.4 MeV/u for A/q = 4.5. This new accelerator, in combination with ISOLDE’s expertise for producing radioisotopes, allows for the re-acceleration of the widest variety of radioactive isotopes worldwide.
The ISOLDE community is now beginning to take advantage of these new opportunities offered at HIE-ISOLDE to peer into the nucleus with a fresh eye. The first two papers from HIE-ISOLDE experiments have recently been published and highlight its potential in two different experiments, both employing the Miniball gamma detector array which can be seen in figure 1.

Figure 1: The Miniball array, used for all the measurements described in this article.
The second paper reports on the Coulomb excitation of even Rn isotopes and is from experiment IS552. Nuclei are known to take on many forms and their shape is dependent on the underlying nuclear structure. E.g. doubly-magic nuclei like 132Sn are spherical, but others cn be prolate (rugby-ball-like) shaped or oblate (pancake-like) deformed. An additional category are nuclei which lack the reflection symmetry of the rugby-ball-like or pancake-like nuclei; these nuclei can take an octupole (pear-like) shape. Nuclei with such an asymmetric static pear-like shape have attracted considerable interest for their role in the search for permanent electric dipole moments (EDMs), as such a moment is expected to be amplified in octupole deformed nuclei. Ra and Rn nuclei have been identified as suitable candidates in the search for EDMs and, when the isotope possesses a static octupole shape, the nuclear Schiff moment due to a non-zero EDM, could be enhanced by a factor of 100-1000. The identification of pear-shaped nuclei is already a specialism of ISOLDE: the observation by Gaffney et al [2] of static octupole deformation in the isotopes of 224Ra and 226Ra was a milestone measurement a few years ago. Now, a new paper builds on this work, and reports on the first spectroscopy of the excited states of even Rn isotopes: 224Rn and 226Rn [3].
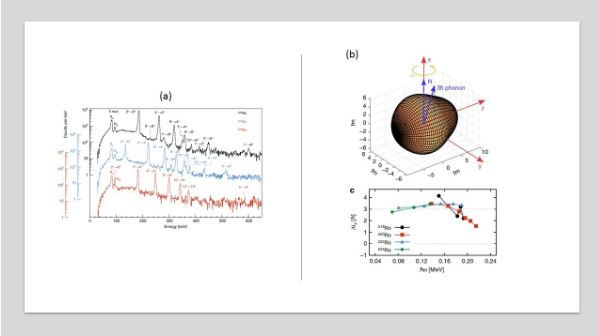
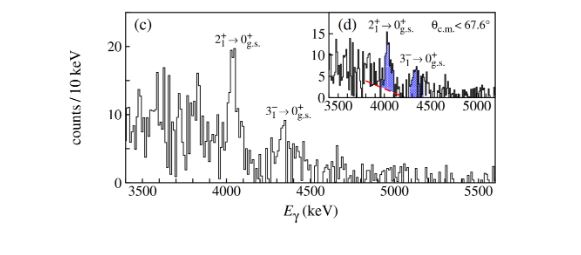
Figure 3: (a) Gamma spectrum from the even Rn isotopes measured at HIE-ISOLDE. (b) Cartoon of phonon coupling in octupole deformed nuclei and the difference in aligned spin for negative- and positive-parity states in 218-224Rn. The dashed line at Δix=0 is the expected value for isotopes possessing static-octupole deformation, indicating that the investigated Rn nuclei do not possess this property.
Rn beams were produced by irradiating a ThC target coupled to a cold plasma ion source. The ions were reaccelerated to 5.08MeV/u and directed towards a target of 120Sn. The re-acceleration – and indeed production – of such heavy ions is unique to ISOLDE worldwide and is among the many aspects where ISOLDE continues to lead the field for radioactive ion beam facilities. A typical spectrum is shown in figure 3 showing the rich gamma spectrum under consideration. Unlike other isotopes – such as 224Ra, which display static octupole deformation [2] – the Rn isotopes do not display this property. Thus, these isotopes are less sensitive for searches for an EDM, contrary to earlier claims.
HIE-ISOLDE has already shown with these first two results the impact which this new accelerator will have, not only in nuclear physics, but beyond. A rich bounty of results is expected from other experiments which have had the opportunity to run before LS2 including the ISOLDE solenoidal spectrometer and the versatile scattering chamber. The first experiments undertaking transfer studies at the high energies offered by HIE-ISOLDE are still being analysed and the user community is eagerly awaiting the restart of the machine after LS2 when it can be exploited to its full potential.
References:
[1] D. Rosiak et al. Phys. Rev. Lett. 121, 252501
[2] Gaffney, L. P. et al. Studies of pear-shaped nuclei using accelerated radioactive beams. Nature 497, 199–204 (2013).
[3] P. Butler et al The observation of vibrating pear-shapes in radon nuclei Nature Communications 10, Article number: 2473 (2019)